

Ein Forschungsteam unter der Leitung von Professor Michel Celliotti von der Eidgenössischen Technischen Hochschule Lausanne (EPFL) hat kürzlich ein neues Machine-Learning-Modell für interatomare Potentiale entwickelt. Dieses verbessert die Effizienz und Anwendbarkeit der Methode in fortgeschrittenen Materialsimulationen deutlich. Ziel der Forschung ist es, die Einschränkungen allgemeiner Modelle hinsichtlich der Trainingsdaten zu überwinden, indem ein neuer Datensatz erstellt und die Architektur des neuronalen Netzes optimiert wird.
Interatomare Potentiale sind mathematische Funktionen, die die Energiebeziehungen atomarer Systeme beschreiben und für die Vorhersage der Stabilität und der Eigenschaften von Materialien entscheidend sind. Bisherige allgemeine Machine-Learning-Modelle für interatomare Potentiale waren aufgrund ihrer Abhängigkeit von spezifischen Systemdaten oder der begrenzten Vielfalt der Trainingsdaten oft nur bedingt anwendbar. Arslan Mazitoff, ein Forscher des COSMO-Labors und Mitglied des Forschungsteams, erklärte: „Mit dem Übergang zu universellen Modellen stehen wir vor dem Dilemma, dass wir zwar die Modelle haben, aber nicht genügend Daten, um sie zu trainieren.“
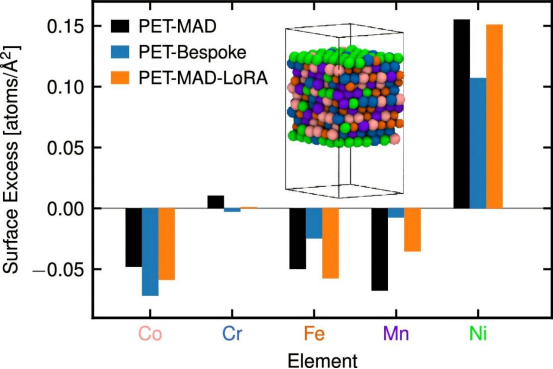
Um dieses Problem zu lösen, veröffentlichte das Team das universelle Modell PET-MAD und den zugehörigen Datensatz zur „großen atomaren Diversität“ in *Nature Communications*. Dieser Datensatz umfasst 85 Elemente und über 95.000 Strukturen und deckt ein breites Spektrum an Materialformen ab, von dreidimensionalen Massenmaterialien bis hin zu Nanoclustern. Alle Daten wurden mithilfe konsistenter Dichtefunktionaltheorie neu berechnet. Mazitoff erläuterte: „Wir haben die Daten so aufbereitet, dass sie kompakter und informationsreicher als frühere Datenbanken sind und sich somit besser für das Training neuronaler Netze eignen.“ Diese Datenbank ist über die Materials Cloud-Plattform öffentlich zugänglich.
Darüber hinaus entwickelte das Team eine Architektur für neuronale Netze, die weniger auf chemisches Vorwissen angewiesen ist und es dem Modell ermöglicht, physikalische Symmetrien während des Trainings selbstständig zu erlernen. Die Studie bestätigte die Effektivität von PET-MAD durch Simulationen von sechs verschiedenen Materialien und demonstrierte eine Genauigkeit, die mit speziell für einzelne Systeme optimierten Modellen vergleichbar ist. Anwendungsgebiete sind die Untersuchung der Ionenleitfähigkeit von Festelektrolyten und die Berechnung des Schmelzpunkts von Galliumarsenid.
Obwohl aktuelle Modelle in Bereichen wie den zugrunde liegenden theoretischen Näherungen und der Beschreibung von Wechselwirkungen über größere Distanzen noch Verbesserungspotenzial aufweisen, liegt ein wesentlicher Beitrag von PET-MAD in der Senkung der technischen und finanziellen Hürden für komplexe Simulationen. Mazitov erklärte: „Wir zeigen, dass PET-MAD durch die intelligente Auswahl und Aufbereitung von Datensätzen die Trainingseffizienz steigern und die Trainingskosten senken kann – typischerweise auf ein Niveau, das selbst für Labore mit kleinem Budget erschwinglich ist –, ohne dabei Genauigkeit, Übertragbarkeit oder Inferenzgeschwindigkeit einzubüßen.“
Weitere Informationen: Autoren Arslan Mazitov et al., Titel: „PET-MAD as a Lightweight Universal Interatomic Potential for Advanced Materials Modeling“, veröffentlicht in *Nature Communications* (2025). Zeitschrifteninformationen: *Nature Communications*